
Introducing the new CuriRx, Inc. Newsletter!

Schedule a Virtual Lunch-N-Learn
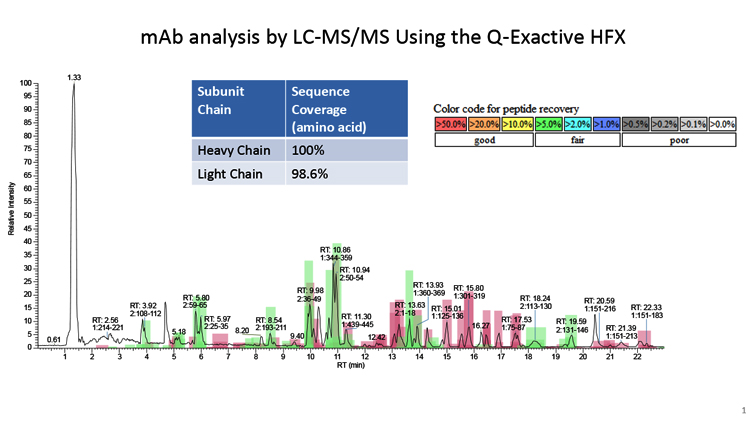
Peptide Mapping Challenges That An Experienced CDMO Can Solve
Peptide mapping using mass spectrometry is a powerful technique for analyzing proteins, with several challenges that must be navigated. CuriRx effectively and efficiently mitigates all of these challenges by applying multiple techniques based on a wealth of experience in biologics characterization. Here are some of the challenges and CuriRx’s novel approach.
Optimized sequence coverage:
- Large hydrophobic regions can be particularly problematic. We solve this by first optimizing sample preparation. Depending on the sample we can decide to denature in a buffer that is amenable to enzymatic digestion, or use stronger conditions followed by robust sample clean-up.
- When needed, CuriRx’s experts will apply multiple and complementary proteases to address smaller, hydrophilic peptides as well as optimizing coverage in CDR regions or other critical quality attributes (CQAs).
Confident Identification Assignments for Peptides and Post Translational Modifications (PTMs)
- CuriRx characterizes biologics using the high-resolution Q-Exactive HF-X mass spectrometer, which is capable of multiple data dependent MS/MS acquisitions throughout a narrow UPLC peak profile. In parallel the full mass spectra of the parent peptide can be acquired at high resolution (>150k).
- This fast acquisition and high-resolution data provide the input for a mass and spectral matching algorithm for peptide identification as well as relative abundance of CQAs. Only those identifications are reported to have confident scoring from the matching algorithm.
Monitoring of Product Quality Attributes and CQAs
- CuriRx takes into account that the type of PTM as well as glycosylations are dependent on the protein sequence as well as bioprocessing conditions.
- Specific product attributes or CQAs can be characterized denovo as well as a targeted approach for a semi-quantitative data analysis.
- We also can characterize and monitor sequence variants and cleavage caused by processing conditions or degradation. We can identify these protein modifications and provide relative abundances by applying appropriate data acquisition parameters, search algorithm inputs, and diligent data analysis.
- Multi-attribute methods (MAM) can be developed to monitor multiple CQAs throughout the development lifecycle of a product.
Incomplete Sequence Coverage
One of the biggest challenges in peptide mapping is achieving complete sequence coverage of the protein of interest.
This is particularly problematic for:
- Highly hydrophobic regions: Peptides containing many aromatic amino acids can be extremely hydrophobic and difficult to retain on typical C18 chromatography columns used in LC-MS workflows.
- Very short peptides: Small, hydrophilic peptides may not be retained well on chromatography columns.
- Complementarity-determining regions (CDRs) of antibodies: These critical regions can be missed entirely if they contain hydrophobic sequences.
Incomplete coverage means potentially missing important post-translational modifications or sequence variants.
Mass Accuracy Limitations
The mass accuracy of the mass spectrometer used can significantly impact the ability to distinguish between similar modifications. Low-resolution instruments such as ion traps may not be able to differentiate between modifications with very similar masses, such as trimethylation (42.04695 Da) and acetylation (42.01057 Da) on lysine residues.
Even with high-resolution instruments, very small mass differences can still pose challenges for unambiguous identification.
Peptide-Spectrum Matching Complexities
Assigning peptide sequences to mass spectra is not always straightforward:
- Isobaric peptides: Different peptide sequences can have identical or very similar masses, making them indistinguishable by mass alone.
- Decoy peptides: False positive matches to decoy database entries can complicate confident peptide identification.
- Peptide length bias: Longer peptides may be more likely to be correctly identified than very short ones.
Protein Inference Issues
Identifying the original proteins from peptide data presents several challenges.
- Shared peptides: Many tryptic peptides are shared across multiple proteins or isoforms, making it difficult to determine the exact protein of origin.
- Leucine/Isoleucine ambiguity: These amino acids are isobaric and cannot be distinguished by mass alone.
- Sample Preparation Challenges: The digestion step can introduce variability and challenges, including:
- Incomplete digestion: Trypsin, the most commonly used protease, may not fully digest all proteins, especially those with resistant regions.
- Alternative proteases: Using proteases like pepsin can improve coverage of hydrophobic regions but may introduce new complexities in data analysis.
Data Analysis and Interpretation
The sheer volume and complexity of data generated in peptide mapping experiments pose significant challenges.
- Heterogeneous data sources: Integrating data from multiple experiments or databases can present challenges due to lack of standardization in protein naming and identification.
- Dynamic range: Detecting low-abundance peptides or modifications in the presence of highly abundant species remains challenging.
To address these challenges, CDMOs like CuriRx are always developing new methodologies and bioinformatics tools. For example, using alternative proteases like pepsin, adding denaturants like guanidine hydrochloride post-digestion, and employing advanced statistical models for peptide and protein identification can all help improve peptide mapping results.
A trusted CDMO partner, combined with evolving mass spectrometry technology and data analysis methods, will continue to advance, many of these challenges may be mitigated, leading to more comprehensive and accurate protein characterization.


